Although many microbial genomes continue to flood the public databases, including different species of Corynebacterium, there has been no corresponding explosion in the knowledge of gene function.
Our transposon-directed insertion site sequencing (TraDIS) technique together with high-throughput approaches will bridge the gap of the high percentage of genes of unknown function in genome sequencing and reveal a high scale genotype-phenotype relationship after comparing genetic fitness. Our primary aim will define essential genes and further identify massively all virulence factors of pathogenic corynebacteria.
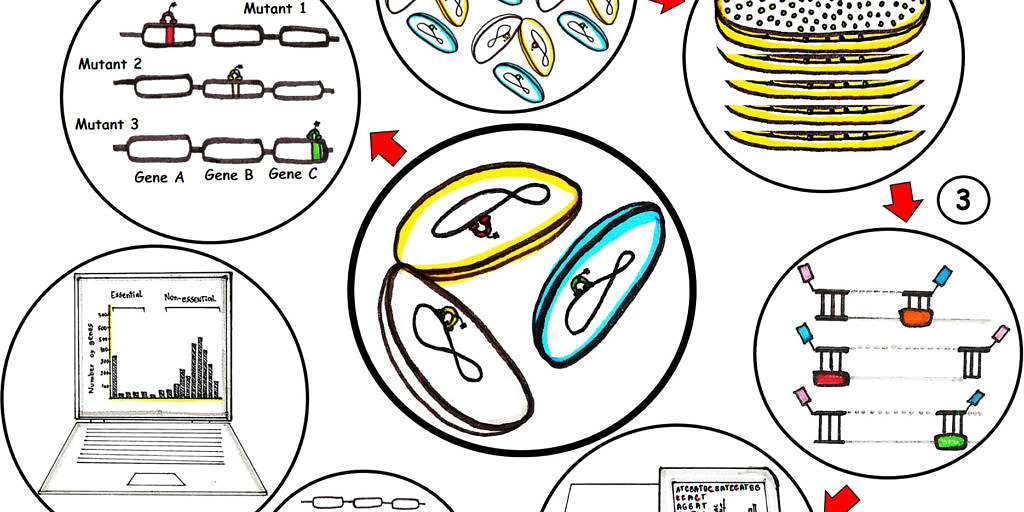
The general idea is to get an improved protocol for a high-density transposon insertion library of at least one million single mutations (1), then cultivate it under a best condition for a greatest bacterial library growth (2) followed by DNA extraction and sample preparation with adaptors ligation at the transposon junctions (3) for the simultaneous sequencing with a next generation sequencer (Illumina platform) (4). Then, the sequence data will be compared with the wild type genome (5) in order to identify the essential and non-essential genes, revealing the genotype-phenotype relationships (6) that are applicable to a range of species.
![]()
Working together the four groups (DKFZ Heidelberg – Dr. Camila Azevedo Antunes Konietzko, FAU – Prof. Burkovski, University of Birmingham – Prof. Henderson and UERJ – Prof. Guaraldi) we have created a high-density transposon insertion library in a non-toxigenic strain of C. diphtheriae isolated from an ill patient. This allowed the identification of all genes that are essential for viability which can be targeted for novel therapeutic strategies. We are currently exploiting this library to identify conditionally essential genes, e.g. those required for resistance to complement dependent killing or those that are required for infecting the host. This information will provide an unprecedented level of understanding of an important human pathogen.